



Investigadores de la Escuela de Medicina de la Universidad Johns Hopkins han identificado un mecanismo regulador dual que protege la integridad mitocondrial mediante las acciones combinadas de Parkin–PINK1 y OMA1. Este estudio, publicado en la prestigiosa revista Nature, revela que estos sistemas de respuesta al estrés controlan la fusión mitocondrial en condiciones fisiológicas normales. La pérdida de ambas proteínas provoca graves anomalías mitocondriales, desarrollo defectuoso y mortalidad temprana en modelos murinos.
Las células responden a estreses patógenos y extrínsecos mediante mecanismos que salvaguardan la salud mitocondrial. Investigaciones anteriores han vinculado las respuestas mitocondriales alteradas con enfermedades neurodegenerativas, insuficiencia cardíaca y síndrome metabólico. Parkin–PINK1 y OMA1 son conocidos sensores de estrés que se activan ante disfunciones mitocondriales.
Parkin, una ligasa de ubiquitina E3, es reclutada a la membrana mitocondrial externa mediante la fosforilación de PINK1, promoviendo procesos de degradación mitocondrial. Por su parte, OMA1 es una proteasa de la membrana interna mitocondrial que inhibe la fusión al escindir el GTPasa de fusión OPA1. Aunque individualmente no son esenciales en condiciones normales, la pérdida combinada de Parkin y OMA1 sugiere una relación compensatoria entre ambos sistemas.
Mecanismos de regulación en modelos murinos
En el estudio, titulado «Regulación dual de la fusión mitocondrial por Parkin–PINK1 y OMA1», los investigadores analizaron la pérdida de sensores de estrés en dieciocho modelos murinos, incluyendo knockouts simples, dobles y triples. Los knockouts homocigotos sistémicos de Parkin y OMA1 presentaron un tamaño corporal reducido, disminución de la actividad locomotora, convulsiones y muerte prematura, con una supervivencia media de 70 días. Fenotipos similares se observaron en ratones knockout de Pink1 y OMA1, subrayando el papel de Parkin–PINK1 en este mecanismo.
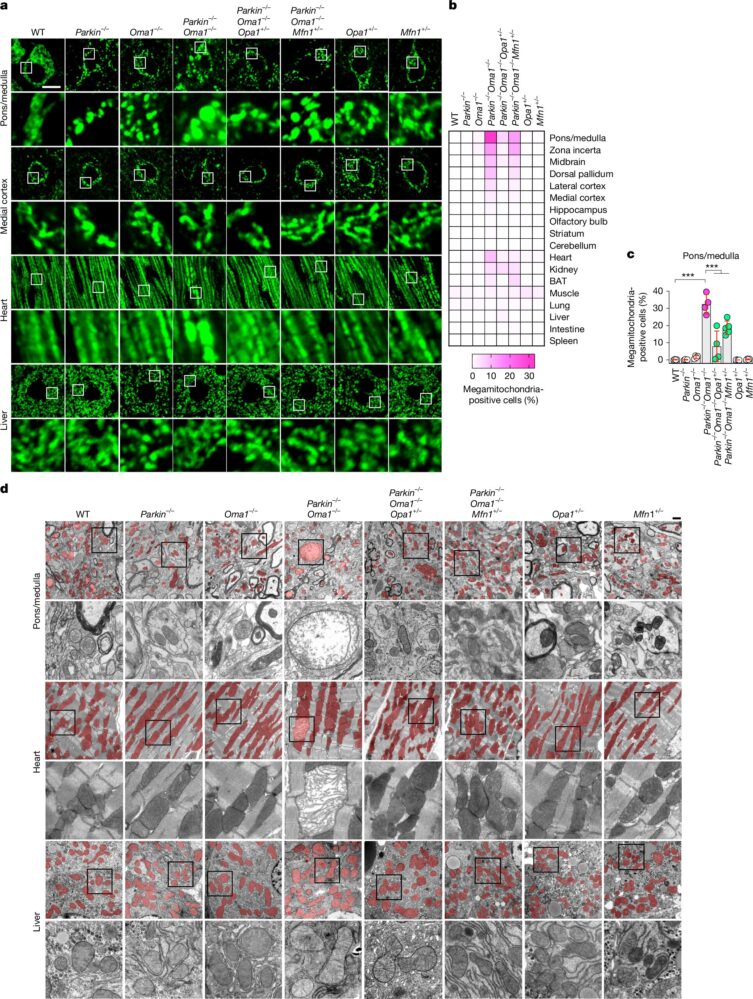
La morfología mitocondrial fue examinada mediante microscopía de inmunofluorescencia confocal láser en diez subregiones cerebrales y ocho órganos principales. Los ratones knockout de Parkin y OMA1 mostraron mitocondrias agrandadas, especialmente en el pons/medula del cerebro y el corazón. La microscopía electrónica de transmisión confirmó la reducción de pliegues de la membrana interna en estas megamitocondrias. La reducción selectiva de OPA1 o MFN1 rescató significativamente las anomalías mitocondriales, la supervivencia y la actividad locomotora, mientras que la reducción de MFN2 no tuvo efecto.
El análisis de secuenciación de ARN del pons/medula reveló la sobreexpresión de genes de respuesta inmune innata en los ratones knockout de Parkin y OMA1. Se detectó ADN mitocondrial (mtDNA) liberado en el citosol, activando la vía STING. Los knockouts triples, que incluían Stinggt/gt, mejoraron la supervivencia y el tamaño corporal, destacando el papel de STING en las respuestas inmunes observadas.
El análisis metabolómico identificó 188 metabolitos sin diferencias significativas entre los tipos silvestres y los knockouts dobles. Las actividades de las enzimas del ciclo del TCA y las tasas de respiración mitocondrial se mantuvieron inalteradas, indicando que la disrupción metabólica no era el defecto primario. En el hígado, los knockouts triples mostraron un aumento exacerbado en el agrandamiento mitocondrial y un deterioro en la mitofagia en comparación con los knockouts de Drp1 solos, correlacionándose estos defectos con niveles elevados de alanina aminotransferasa en suero, lo que indica daño hepático.
El análisis de neuronas motoras mostró una reducción en el conteo de neuronas en la médula espinal torácica y lumbar de los knockouts dobles, lo que explica los déficits locomotores. Las neuronas dopaminérgicas en la sustancia negra y los niveles de dopamina en el cuerpo estriado no se vieron afectados.
Los resultados experimentales demuestran que Parkin–PINK1 y OMA1 controlan tanto eventos de la membrana externa como interna, compensándose mutuamente cuando es necesario y colaborando para prevenir la fusión mitocondrial excesiva, manteniendo la estructura mitocondrial y la integridad del genoma. La pérdida de ambas vías interrumpe la dinámica mitocondrial, desencadenando respuestas inmunes y disfunción de órganos, lo que indica que el mecanismo regulador dual es crítico para el desarrollo, la fisiología y la supervivencia animal.



