La química computacional ha experimentado avances significativos en las últimas décadas, transformando la forma en que los científicos abordan el diseño y la caracterización de materiales. Investigadores del Instituto Tecnológico de Massachusetts (MIT) han desarrollado un nuevo enfoque que combina la teoría cuántica con técnicas de aprendizaje automático, prometiendo mejorar la precisión y la eficiencia en la predicción de las propiedades electrónicas de las moléculas.
Históricamente, la creación de nuevos materiales ha sido un proceso arduo. Desde hace más de mil años, científicos como Tycho Brahe, Robert Boyle e Isaac Newton se aventuraron en la alquimia, buscando convertir elementos como el plomo o el mercurio en oro. Sin embargo, el desarrollo de la tabla periódica de elementos ha proporcionado un marco más sólido para entender las propiedades de los materiales, permitiendo a los investigadores centrarse en combinaciones químicas más viables.
Avances en química computacional
El estudio reciente liderado por Ju Li, profesor de ingeniería nuclear y ciencia de materiales en el MIT, ha sido publicado en Nature Computational Science. Este trabajo se basa en la teoría de funcionales de densidad (DFT), un método cuántico ampliamente utilizado para calcular la energía total de moléculas y cristales. Aunque la DFT ha demostrado ser eficaz, presenta limitaciones en términos de precisión y en la información que puede ofrecer, ya que solo calcula la energía mínima de un sistema molecular.
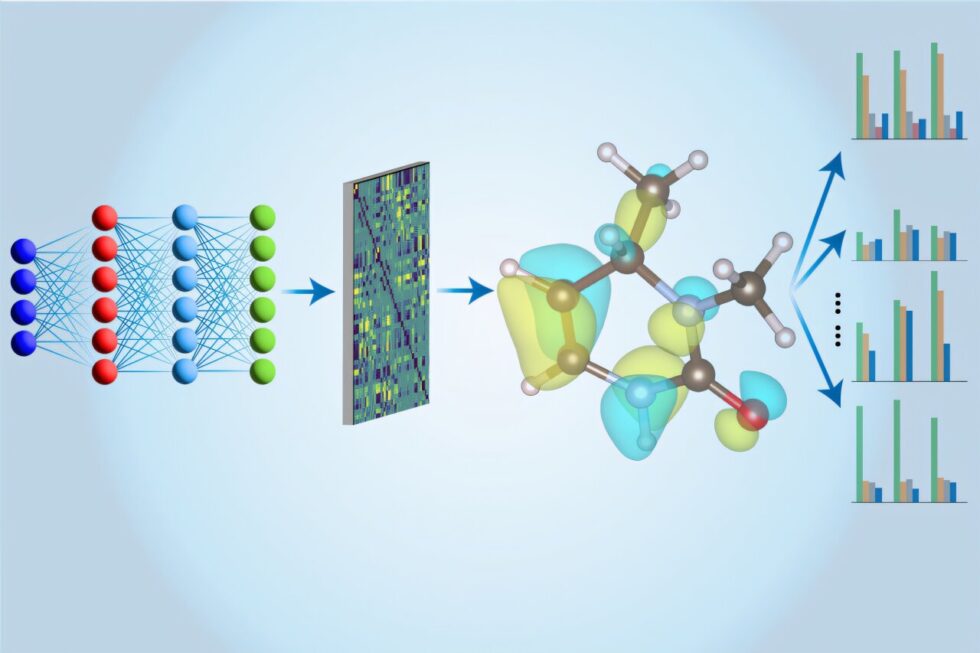
El equipo de Li ha decidido utilizar una técnica alternativa conocida como teoría de cúmulo acoplado (CCSD(T)), considerada el estándar de oro en química cuántica. A pesar de su alta precisión, los cálculos con CCSD(T) son computacionalmente intensivos, lo que limita su aplicación a moléculas pequeñas. Para superar este obstáculo, los investigadores han implementado un modelo de red neuronal que permite realizar estas complejas simulaciones de manera mucho más rápida y eficiente, extrayendo información sobre diversas propiedades electrónicas más allá de la energía total.
El modelo, denominado «Multi-task Electronic Hamiltonian network» (MEHnet), no solo evalúa la energía de las moléculas, sino que también proporciona datos sobre momentos dipolares y cuadrupolares, polarizabilidad electrónica y la brecha de excitación óptica. Esta última propiedad es crucial para entender cómo los materiales interactúan con la luz, lo que tiene implicaciones directas en el diseño de nuevos compuestos ópticos y fotónicos.
Los resultados iniciales han mostrado que el modelo MEHnet supera a la DFT en precisión y se alinea estrechamente con los resultados experimentales. Investigadores externos, como Qiang Zhu de la Universidad de Carolina del Norte, han elogiado la capacidad del modelo para entrenarse eficazmente con conjuntos de datos reducidos, lo que es un avance notable en la sinergia entre la química computacional y el aprendizaje profundo.
Este nuevo enfoque se ha aplicado inicialmente a elementos no metálicos como el hidrógeno, carbono, nitrógeno, oxígeno y flúor, y se prevé que su uso se extienda a elementos más pesados como el silicio, fósforo y azufre. La versatilidad del modelo permite abordar moléculas cada vez más grandes, potencialmente alcanzando miles de átomos, lo que representaría un cambio significativo en la capacidad de análisis en química computacional.
El futuro de esta investigación apunta a aplicaciones prácticas en el diseño de nuevos polímeros, materiales semiconductores y compuestos para baterías, áreas que son de vital importancia en el contexto actual de sostenibilidad y eficiencia energética. La ambición de Li y su equipo es abarcar toda la tabla periódica con un nivel de precisión comparable al de CCSD(T), pero a un costo computacional inferior al de la DFT, lo que podría revolucionar el campo de la química, la biología y la ciencia de materiales.